

VCP - Developing a neuroprotective drug therapy to slow vision loss in inherited retinal disease
To date, almost all forms of retinal degeneration (RD) remain untreatable, resulting in visual impairment and blindness. Based on strong experimental data, we have reasons to believe that disturbances in proteostasis and energy depletion are common features in RD. Given the high energy demand of photoreceptors, chronic disruptions in their cellular protein and metabolic homeostasis
will inevitably lead to dysfunction, energy depletion, and, eventually, cell death. We have identified “Valosin-containing protein” (VCP), an ATPase essential for the high energy-consuming process of degradation of misfolded proteins, as a key therapeutic target. Notably, rebalancing proteostasis by inhibiting VCP with three 1 st generation VCP inhibitors improved photoreceptor viability and function in three genetically distinct animal models for RD (RHOl256del, RHOP23H, Pde6brd10). Now, we want to advance this preclinical proof of concept to identify and validate a superior VCP-targeting drug for future clinical application in man. This approach is based on a small molecule compound library of 2 nd generation VCP inhibitors with advanced physicochemical, pharmacological, and biological qualities. Focusing on inhibition of our highly validated target, VCP, the objective of this proposal is to build a diligent basis and rationale for a mutation-independent pharmacological approach to treat inherited RD by identifying a drug as well as indication criteria that can serve as a basis for entering a clinical phase I/II study. Including models for dominant and recessive RD, we will investigate how pharmacological inhibition of VCP counteracts the loss of photoreceptors affected by RD-type diseases that are not directly linked to misfolding of the respective protein.
The success of this project will generate superior drugs and crucially advance the knowledge about which forms of RD respond most efficiently to pharmacological inhibition of VCP and which patients might benefit most in the future. Target VCP will pave the way toward bringing this promising therapeutic concept as a new medical therapy to patients.
EYE-RISK
Using a systems medicine approach, EYE-RISK aims at identifying risk factors, molecular mechanisms and therapeutic approaches for the complex eye disease age-related macular degeneration (AMD). AMD is the leading cause of blindness in European countries. The disease is characterized by the degeneration of the central part of the retina called macula. This part is needed for central vision and is crucial for tasks such as reading, driving, recognition of faces and color vision. The frequency of AMD in the general population increases sharply after an age of 65 years up to a prevalence of 10% after an age of 80 years. The risk to develop AMD is jointly determined by age, lifestyle and also by the individual genetic background. At the current state, AMD is an incurable disease.
The EYE-RISK project utilizes epidemiological data describing clinical phenotype, molecular genetics, lifestyle, nutrition, and in-depth retinal imaging derived from existing longitudinal European epidemiological cohorts and biobanks to provide three major insights needed for long-lasting prevention and therapy of AMD:
- Development of robust algorithms utilizing genetic and non-genetic risk factors to identify personalized risks of developing advanced wet and dry AMD.
- Identification of novel biomarkers for further stratification of disease risks.
- New insights from 1) and 2) will be used to elaborate preventive medical recommendations for high-risk subgroups of AMD patients.
- Identification of molecular drivers / biological pathways relevant for onset and progression of advanced AMD that will be used to identify and validate new therapeutic targets.
More information on the website of the project www.eyerisk.eu
RD-Cure and Coroideremia Gene Therapy Trials
- Gene Therapy Trial for Achromatopsia (For patients with mutations in the CNGA3 gene)
- Gene Therapy Trial for Retinitis Pigmentosa (For patients with mutations in the PDE6A gene)
With an estimated prevalence of 1:4 000 and approximately 1 million affected patients worldwide, retinal dystrophies are a major cause of visual disability. The different forms of retinal dystrophies can be attributed to mutations in more than 100 genes. To date, no cure is available for routine and general application. Researchers are currently investigating the potential for gene therapy in treating retinal dystrophies. Up to now, gene therapy in humans is only performed within clinical trials.
The Centre for Ophthalmology of the University Hospital Tübingen collaborates with partners of the Ludwig-Maximilian-University in Munich and the Columbia University in New York on the clinical translation of gene therapy for retinal dystrophies. To achieve this goal, a consortium of clinicians and researchers with complementary expertise has been assembled and started work in October 2012. The project is funded by the Tistou & Charlotte Kerstan Foundation.
From the group of retinal dystrophies, the RD-Cure-Consortium has selected two genetic subforms – CNGA3-associated Achromatopsia (Study 1) and PDE6A-associated Retinitis Pigmentosa (Study 2) – for which a therapeutic approach for application in clinical trials shall be developed. Both studies will make use of a supplementary gene therapy targeting retinal cells. Supplementary gene therapy aims at a restoration of insufficient or lacking gene function as a result of a pathogenic mutation. Recombinant adeno-associated viral vectors will be used for retinal gene transfer.
The RD-Cure project is currently in the preclinical phase. Partners of the consortium have already performed proof-of-concept studies in mouse models. Toxicological and biodistribution studies are currently performed. Clinical evaluation of patients eligible for therapy is in progress. The clinical phase for Study 1 is expected to start in 2015, while Study 2 is planned for 2017.
More information on the website of the project: www.rd-cure.de
DRUGSFORD: Drugs for Retinal Degeneration
The DRUGSFORD project aims at finding new ways to counteract hereditary photoreceptor degeneration. DRUGSFORD is supported by the EU 7th framework programme and has started on 1st September, 2012.
The retina sits in the back of our eyes and is a tissue that we depend on for our ability to see. Within the retina, the photoreceptor cells are responsible for capturing light and for transforming it into messages that can be sent to and interpreted by the brain. Photoreceptors are nerve cells, and will not be replaced when lost, which means that if they die, the retina loses its ability to capture light forever, with dramatic consequences for vision.
The DRUGSFORD consortium consists of two biotech companies and three academic research groups. The German company BIOLOG (Bremen) is a world leader in developing and producing compounds that interact with various cGMP-governed cellular reactions. The Dutch company to-BBB (Leiden) has developed specialized drug delivery systems that help pharmaceutical substances pass across the blood-brain-barrier. The combined expertise of these two companies allows the project to come up with novel drugs and ways to deliver them across the blood-retina-barrier, so that they can reach the photoreceptors. The academic research groups are connected to Valeria Marigo (University of Modena and Reggio Emilia, in Modena, Italy), Per Ekström (Lund University, in Lund, Sweden) and François Paquet-Durand (Eberhard Karls University, in Tübingen, Germany), and have already previously collaborated regarding photoreceptor degeneration mechanisms and the protective effect of treatments with various substances. The academic groups will test the drug and delivery systems for their protective effects on diseased photoreceptors and retinas.
Together, the DRUGSFORD consortium members aim at having a photoreceptor protective drug and delivery system ready for initial clinical trials by the finalization of the three-year project period.
More information on the website of the project: www.drugsford.eu
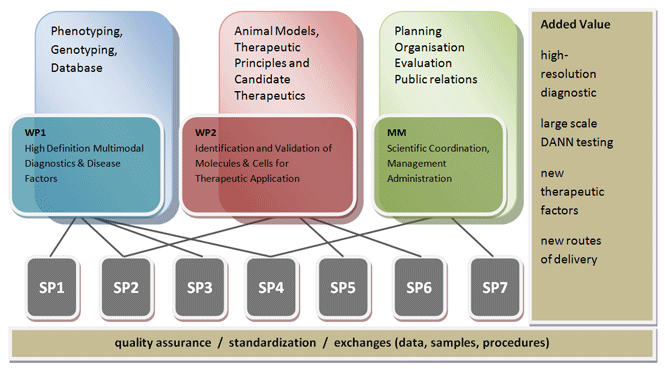
HOPE: Hereditary Retinal Disorders: From Patients towards Therapies
Hereditary retinal disorders (HRDs) represent a very heterogeneous group of blinding diseases affecting about 30.000 people in Germany.
HRDs do not only cause tremendous socio-economic costs but also severely impair life quality for a long period of time. In Germany, clinical patient care and the availability of integrated diagnostics for HRDs is not well established and most importantly, there is currently no effective cure available, although the accessibility of the retina to both, clinical investigations and surgical intervention and its immuneprivileged status favour the development of rational therapies and improved clinical markers.The HOPE consortium has started its work in April 2009. Profound progress with respect to our principle objectives and tasks was made since then.
The HOPE consortium has its focus on (1) the improvement of clinical phenotyping by integrating multiple functional and morphological parameters, (2) the development of qualitycontrolled high-throughput DNA diagnostics for HRD, (3) the establishment of a national network of clinical experts in HRDs, (4) the identification of genetic modifiers that govern penetrance and disease expression, (5) the identification of neurotrophic factors with therapeutic potency for HRDs, (6) the adaptation of encapsulated cell technology (CellBeads) for the delivery of therapeutic compounds in the eye, and (7) the monitoring of the biocompatibility, survival and therapeutic efficacy of CellBeads in animal models for HRDs.
More information: http://www.rd-hope.de/
Involved partners from the Institute: Kohl, Muehlfriedel, Seeliger, Ueffing, Wheeler-Schilling, Wissinger, Zrenner
SYSCILIA: A systems biology approach to dissect cilia function and its disruption in human genetic disease
The aim of SYSCILIA is to identify the molecular mechanisms characterizing cilium function, and the discrete perturbations associated with dysfunction caused by mutations in inherited ciliopathies, applying a systems biology approach. Our overall objectives are to establish a paradigm for studying and modelling complex eukaryotic systems, to understand how system perturbation contributes to the modulation of clinical phenotypes, and to provide a better understanding of ciliary processes in biology and their associated diseases.
Component 1 - “Defining the elements and variables of ciliary systems”
Component 2 - “Modelling the variables of ciliary systems”
Component 3 - “Assessing and manipulating the variables of ciliary systems”
Component 4 - “Applying ciliary systems to human health towards improved diagnostics and therapy”
Component 5 - “Management”
Involved partners from the Institute: Ueffing
Download the overview poster: http://www.syscilia.org/docs/Poster_SYSCILIA_CILIA2012_Roepman_v1_A4.pdf